Зміст
- Роль лізосом у розвитку захворювання
- Поширеність
- Хто з батьків «винен» у спадкуванні?
- Симптоми
- Проблеми діагностики
- Ускладнення
- Лікування
- Прогноз
Хвороба Фабрі — дуже рідкісне, що передається у спадок захворювання. З-за генної мутації клітинних лізосомах зникає важливий фермент α-галактозидаза. В нормі він займається переробкою зайвих жирів (сфінголіпідів) непотрібних організму. При захворюванні ліпіди накопичуються у всіх клітинах, що поступово призводить до їх руйнування.
Захворювання відноситься до групи лізосомальних хвороб накопичення. Перший опис дали майже одночасно два лікаря дерматолога: німець Джон Фабрі (1860-1930) і англієць Вільям Андерсон. Тому патологія у деяких країнах іменується хворобою Андерсона-Фабрі. Автори помітили істотну зв’язок шкірних проявів при ангиокератоме з виділенням білка в сечу, варикозним розширенням вен, деформацією пальців.
Роль лізосом у розвитку захворювання
Всі клітини мають свої внутрішні механізми для усунення зайвих молекул. Лізосоми – це внутрішньоклітинні структури у вигляді найдрібніших бульбашок. Вони мають власну оболонку і наповнені ферментами гидролазами. Всередині підтримується кисле середовище, необхідна для «роботи» ферментів. Налічується близько 40 видів гідролаз. Кожна «курирує» певну біохімічну реакцію. У разі хвороби Фабрі – це α-галактозидаза.
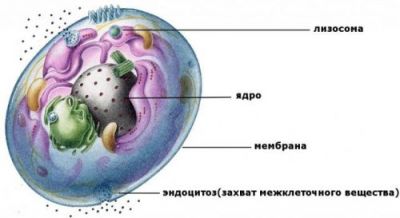
Лізосоми беруть активну участь в розщепленні, тільки якщо в їх структурі не є порушень
Порушена робота ферменту призводить до накопичення сфінголіпідів, у зв’язку з чим з’являються несприятливі для організму наслідки. Жири в організмі людини утворюють різні сполуки. Сфинголипиды відрізняються від звичайних фосфоліпідів відсутністю гліцерину у своїй структурі. Цей вид жирів входить до складу оболонки нервових клітин (нейронів). Накопичення в лізосомах викликає збільшення розмірів органів, аномалії в кісткової тканини, ураження легень, нирок та іншої патології.
Величина накопичених жирів розподіляється в пропорції аналогічної нормального вмісту в тканинах.
Поширеність
Поширеність хвороби Фабрі серед новонароджених коливається, за різними даними, від 1 випадку на 40 000 дітей, народжених до 1 на 120 000. Вважається, що ця патологія займає друге місце після хвороби Гоше.
Будь-якого зв’язку з расовими особливостями не помічено. Найбільш типові ознаки ураження одного органу і легкий перебіг захворювання.
Хто з батьків «винен» у спадкуванні?
Встановлено, що в 95% випадків діти отримують дефектний ген від матері або батька. При цьому хлопчики – тільки від матері, дівчинки – як від матері, так і від батька. Це доводить зв’язок мутантних генів з підлогою, а саме з Х-хромосомою:
- у чоловіків одна Х-хромосома, якщо в ній дефект, то вона передається за законами спадкування тільки дочки;
- у жінок дві аналогічних хромосоми, при ураженні однієї з них ймовірність передачі патології становить 50%, в які входять і сини, і дочки.
У 5% хворих не встановлена зв’язок з генами батьків. Мутація в гені GLA, відповідальному за фермент α-галактозидазу, відбувається вперше на рівні організму дитини. Це має значення в діагностиці.
Встановлені особливості перебіг хвороби Фабрі, отриманої по материнській лінії:
- симптоматика виражена помірно;
- пізніше початок;
- повільно прогресуючий характер;
- порівняно легкі зміни в органах і тканинах.
Симптоми
Симптоми хвороби Фабрі можуть виявлятися у дитини в ранньому дитячому віці і у дорослої людини. Прийнято розрізняти:
- класичне протягом з наявністю кількох ознак;
- некласичне (атиповий).
Ознаки захворювання дуже різноманітні. Зовнішній вигляд – наближається до класичного у віці 12-14 років. Більш типовий для хлопчиків:
- надбрівні дуги і лоб виступають вперед;
- перенісся западає;
- нижня щелепа збільшена;
- великий рот і широкі губи.
На фото хвороби Фабрі неспеціалістові складно помітити зміни пропорцій обличчя
Ендокринологів такі зміни зовнішності нагадують акромегалію (хвороба, викликану підвищеною продукцією соматотропного гормону).
Больовий синдром – вважається найбільш раннім і частим проявом хвороби. Можливі 2 типу:
- нейропатіческая – у вигляді печіння, поколювання в ступнях і долонях;
- кризи – раптові різкі болі пекучого характеру, максимально локалізуються в стопах і долонях, але можуть поширюватися на інші ділянки тіла, тривають від декількох хвилин до тижня, виснажують пацієнтів, інколи супроводжуються підвищенням температури.
 Ознаки захворювання нирок
Ознаки захворювання нирок
Кризи провокуються зміною погодних умов, перегрівом тіла, стресовими ситуаціями, фізичним навантаженням, втомою. Порушене потовиділення – викликано пошкодженням нервових волокон і клітин потових залоз, відсутність пітливості призводить до непереносимості спеки, оскільки організм перегрівається.
Швидка стомлюваність, слабкість, погана переносимість фізичного навантаження сильна слабкість призводить до неможливості рухової активності, навіть при помірних навантаженнях пацієнти відчувають втому, перегрівання, можлива поява нападоподібний біль. У тяжких випадках людина абсолютно безпорадний.
Шкірні прояви – згадаймо, що хвороба Фабрі описана за наявності ангиокератом. Це дрібні синюшні висипання на губах, пальцях рук і ніг, в області пупка, зовнішніх статевих органах, промежині, на сідницях, колінах, ліктях, частіше в місцях розтягування шкіри. Вони можуть збільшуватися в діаметрі до 10 мм з дорослішанням пацієнта. Відносяться до ранніх проявів патології, але не вважаються обов’язковою ознакою, оскільки відомо досить захворювань з аналогічними висипаннями.
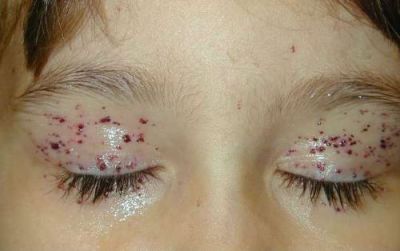
Ангиокератомы можуть локалізуватися в області верхніх повік
Симптоми ураження серця – пацієнти скаржаться на болі за грудиною, що нагадують стенокардію, по суті своїй вони викликані зміною коронарних судин, звуженням їх просвіту. Діагностується гіпертрофічна кардіоміопатія (частіше необструктивний типу, рідко – з обструкцією, ураженням верхівки серця). З’являються різні аритмії. Від порушення діяльності серця пацієнти помирають. У половини хворих виявляється пролапс мітрального клапана, який поєднується з розширенням аорти.
Кератопатия – ознака змін на рогівці виявляють при огляді офтальмолога, вона називається «лійкоподібної», не впливає на зір.
Зміни слуху у хворих виникає «дзвін у вухах, шум», часто прогресує падіння слуху. З боку нервової системи спостерігаються типові симптоми ішемічного інсульту або транзиторних атак (головні болі, запаморочення, односторонні парези паралічі кінцівок, порушення шкірної чутливості). Ранні інсульти у молодих людей без поширеного атеросклерозу завжди викликають підозру на хворобу Фабрі.
Психічні порушення – зміни психіки є наслідком перебігу захворювання з постійними болями і іншими ознаками. Пацієнти відрізняються підвищеною тривожністю, недовірою, мінливим настроєм, невпевненістю, нездатні виконувати точну роботу.
Порушення з боку нирок – характерні для хворих дітей, багатьох жінок і чоловіків. Зміни в нирковій тканині пов’язані з відкладенням ліпідів у эндотелиальном шарі судин клубочків. Важливо, що вони призводять до необоротної ниркової недостатності до 40-річного віку пацієнта і летального результату захворювання. Початкові ознаки проходять непоміченими.
На відміну від ниркової недостатності при хронічних ураженнях паренхіми у пацієнтів немає гіпертонії, білок у сечі визначається у мінімальній кількості, креатинін в крові в нормі. Причину відмови функції нирок можна встановити тільки при серйозному обстеженні.
Суглобовий синдром – біль та припухлість суглобів спостерігається у 1/3 дітей. У дитини виникають болі в м’язах при ходьбі, рухах. Найбільш частий діагноз – ревматизм. Тромбоемболії глибоких периферичних вен кінцівок – викликані збільшенням здатності тромбоцитів до склеювання (агрегації), підвищенням рівня тромбоглобулинов в плазмі. Нерідкі випадки емболії легеневої артерії та портальної вени.
Проблеми діагностики
У зв’язку з рідкістю і багатогранними ознаками, хвороба Фабрі важко діагностувати в звичайних лабораторіях. Пацієнти лікуються у різних фахівців з підозрами на іншу патологію. До правильної постановки діагнозу проходить в середньому 12 років. А це важливий упущений момент лікування.
У РФ ферментними і генетичними захворюваннями займаються в Лабораторії спадкових хвороб обміну речовин при медичній академії і в Лабораторії молекулярно-генетичної діагностики Центру здоров’я дітей в Москві.
Для повного обстеження проводять:
- розпитування про прояви захворюваності в родоводі матері і батька;
- дослідження рівня ферменту галактозидази в крові (для жінок норма не вважається ознакою відсутності захворювання);
- аналіз виділеної ДНК з будь-якого біологічного матеріалу;
- вимірювання рівня ферменту в тканинах плода у разі виявленої патології у матері (пренатальна діагностика) для з’ясування вірогідності успадкування хвороби дитиною;
- біопсію тканини нирок для виявлення гістологічних змін в лізосомах;
- електрокардіографію та ехокардіографію з метою виявлення порушень діяльності серця;
- магниторезонансную томографію головного мозку.

Визначення наявності мутацій – один з методів діагностики
Особливості лабораторної діагностики
Найбільш чутливими тестами є:
Для пренатальної (допологової) діагностики захворювання плода досліджують галактозидазную активність в клітинах з амніотичної рідини.
Ускладнення
Відсутність доступної діагностики призводить до неправильного лікування, пізнього виявлення хвороби. У пацієнтів у важкій стадії з’являються:
- ниркова недостатність з протеїнурією і незворотними змінами в паренхімі органу;
- інсульти з неврологічними наслідками;
- порушення серцевої діяльності у вигляді інфаркту міокарда, гіпертрофії лівого шлуночка, аритмії.
Лікування
Людей, з підозрою на хвороба Фабрі, госпіталізують у спеціальні центри, які профільовані для лікування і досліджень патологій накопичення та ферментної недостатності. Терапія спрямована:
- на заміщення відсутньою α-галактозидази;
- боротьбу з негативними наслідками і змінами в органах і системах.
В якості замісної терапії пацієнти отримують препарати з α – і β-галактозидазой
У РФ аптечна мережа отримує для лікування рідкісних хвороб 2 види замісних препаратів. Реплагал (по хімічній структурі містить агалсидазу-альфа) – проводиться з клітин людини, вводиться внутрішньовенно на фізрозчині, крапельно повільно (протягом 40 хвилин) кожні 2 тижні. Дозування розраховується за віком і вагою пацієнта. Протипоказаний дітям до 7 років (немає досліджень), вагітним і годуючим жінкам. Якщо хворий перебуває на гемодіалізі, доза не змінюється.
До частих негативних реакцій належать:
- головний біль;
- запаморочення;
- спотворення смаку;
- тремор;
- сльозотеча;
- шум у вухах;
- «припливи до обличчя;
- прискорене серцебиття;
- підвищення артеріального тиску;
- нудота;
- пронос;
- озноб з підвищенням температури;
- болі в м’язах;
- припухлість суглобів;
- посилення слабкості.
Фабразим (агалсидаза-бета) – отримують з ДНК клітин китайського хом’ячка, відрізняється вираженими алергічними проявами у пацієнтів, при внутрішньовенному введенні рекомендується бути готовими до проведення протишокових заходів. Поки немає відомостей про взаємодії цих препаратів з іншими ліками.
Теоретично не рекомендується змішувати їх в одному флаконі, застосовувати на фоні лікування пацієнта:
- Хлорохіном;
- Бенохином;
- Аміодароном;
- Гентаміцином.
Очікується ризик пригнічення активності ферментних препаратів. При вираженому больовому синдромі призначаються протисудомні засоби, анальгетики. Болі в суглобах допомагають зняти нестероїдні протизапальні засоби. Прояви ниркової недостатності купіруються введенням ферментів. При серцевих симптомах додатково використовуються коронаролітики, протиаритмічні препарати.
Прогноз
Сучасні дослідження показують успішне лікування хвороби Фабрі з допомогою замісної ферментної терапії. Контрольні аналізи довели нормалізацію рівня в крові і призупинення розвитку захворювання. Якщо лікування розпочато вчасно, прогноз цілком сприятливий.
Жінок з хворобою Фабрі спостерігають у період вагітності. Загрози для стану здоров’я матері та дитини хвороба не представляє, хоча спостерігалися випадки посилення протеїнурії, гіпертензії, психічних змін під час вагітності та післяпологовому періоді.
Головна проблема захворювання – складності діагностики і дорожнеча лікування (1 флакон Реплагала у 3,5 мл коштує від 125 до 168 тис. руб.). Відсутність своєчасної терапії призводить до ранніх ускладнень, зниження тривалості життя. У дитячому і юнацькому віці пацієнти не здатні вчитися, виконувати фізичну роботу, вести повноцінне життя.